吸附能的计算
大约 1 分钟
吸附能的计算
Todo
影响因素
slab 模型在 Z 方向
- slab 的厚度
- 真空层厚度
一方面影响计算量的大小,另一方面,对于不同的体系,我们需要不同厚度的 slab 模型来保证计算的准确性。例如:对于金属体系来说,越开放的表面往往需要更多的层数。测试或参考他人成果。
slab 模型在 XY 方向
- 表面大小:影响覆盖度,计算的工作量。
- 吸附位点:top(t),bridge(b),fcc(f) 和 hcp(h)。
- 吸附物种与表面结合情况:不同的分子构型?用什么原子?哪一部位和表面接触?初始猜测的键长多少? 等等
初始构型获取
- 查数据库
- 查文献
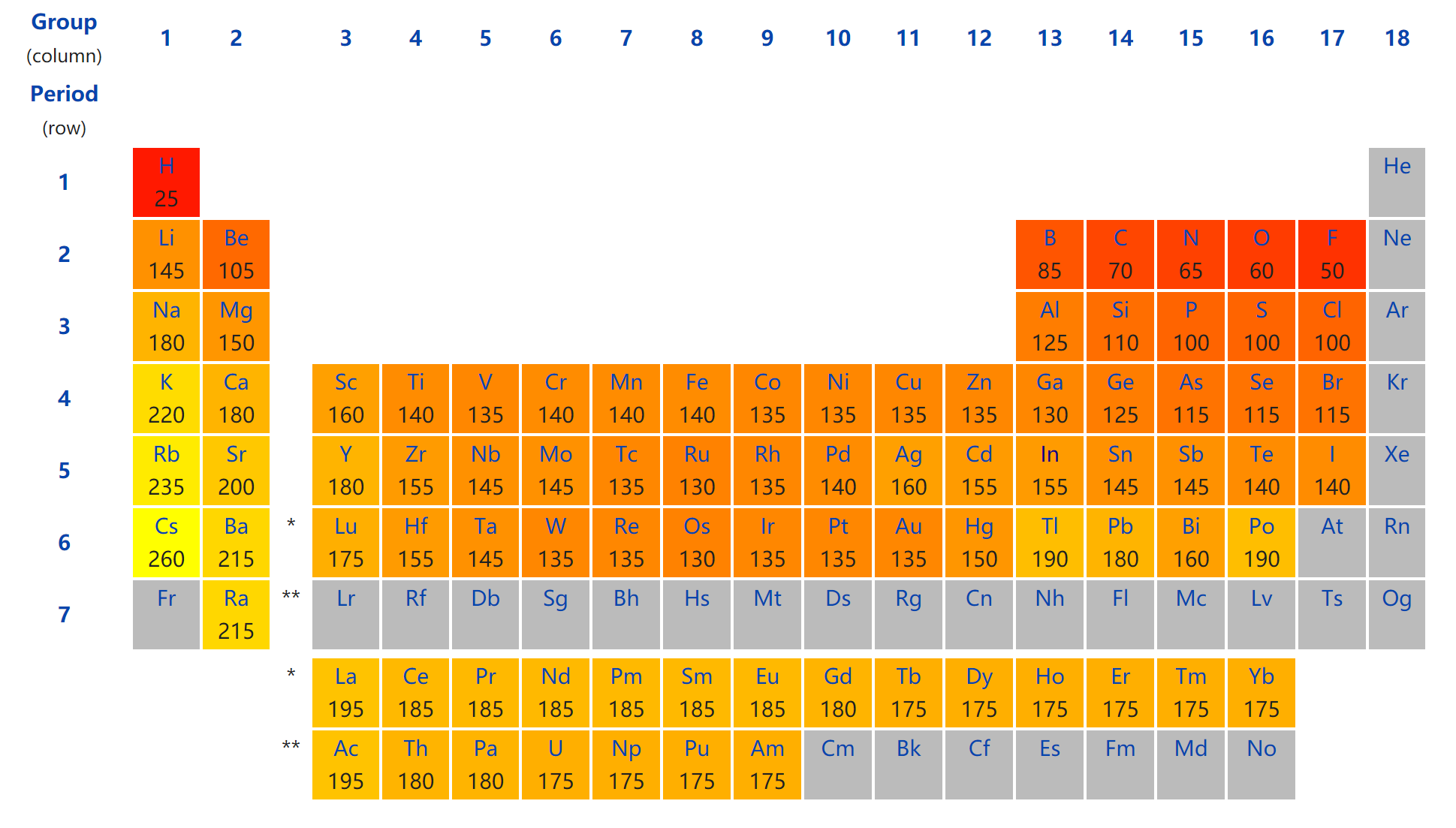
- 自己估算:依据原子半径,两原子成键,键长小于两者之和。下图 😃
- 初算:用一个小模型,简单算一下,得到一个合理的键长。比如 O 吸附在 Cu,直接优化气相中 Cu-O 双原子分子的结构。Ex54 简单粗暴地获取初始构型(一)| Learn VASP The Hard Way (bigbrosci.com)